Hemophilia Genotyping Results from the My Life, Our Future Project
Objective:
My Life, Our Future (MLOF) is a national project directed by a partnership formed to: 1) conduct wide-scale genetic testing of the U.S. hemophilia community, thereby increasing the rate of patient testing above the currently estimated 20% and allowing carrier detection; 2) establish a repository of associated samples and data to support scientific discovery and treatment advances including informing inhibitor risk and disease severity.
Methods:
A multi-sector partnership was formed to make hemophilia genotype analysis available to the U.S. community. The National Hemophilia Foundation (NHF) educates consumers and supports recruitment. The American Thrombosis and Hemostasis Network (ATHN) provides hemophilia treatment center (HTC) provider education, a secure infrastructure for data collection, and point of access for research proposals. HTCs enroll patients, obtain samples and provide clinical results to patients. Puget Sound Blood Center (PSBC) serves as the central genotyping laboratory and sample repository. Biogen Idec provides scientific collaboration and initiative support.
A pilot study involving 11 HTCs was successfully completed in 2013, and patients continue to be enrolled at those and additional sites. Genotyping is performed through an initial screen by Next Generation sequencing of extracted DNA using a molecular inversion probe-based capture strategy. FVIII and FIX mutations are confirmed in the CLIA-certified PSBC hemophilia genomics laboratory using a separate DNA sample. A clinical laboratory report is returned to the HTC and results transmitted into the ATHN Clinical Manager database accessible only to the patient’s HTC providers. For patients who give informed consent, coded data and samples are stored in a research repository, which can be linked to coded clinical data from the ATHNdataset for future research applications. NHF’s national and local chapter educational programs increased awareness and educated families about MLOF.
Summary:
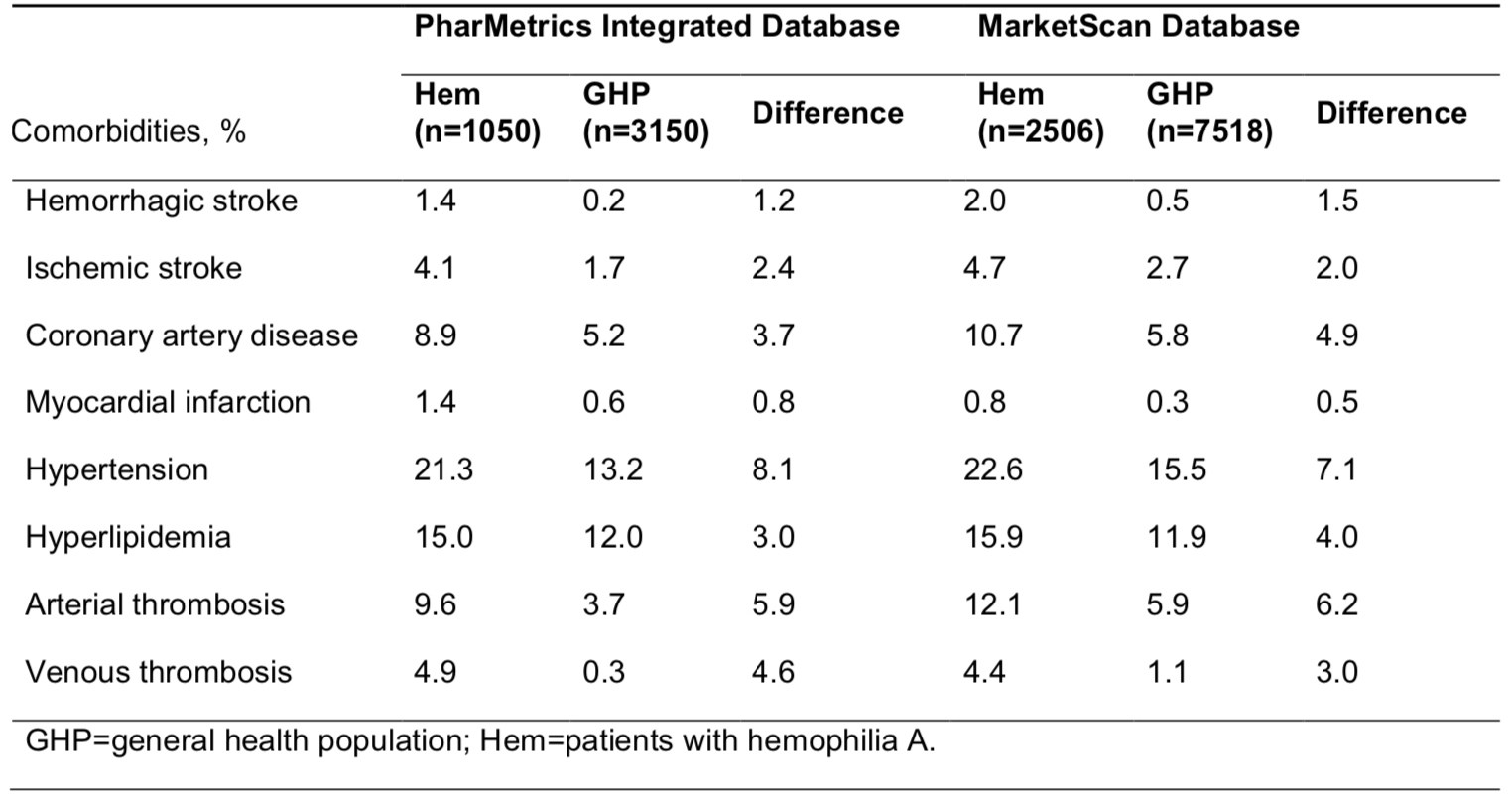
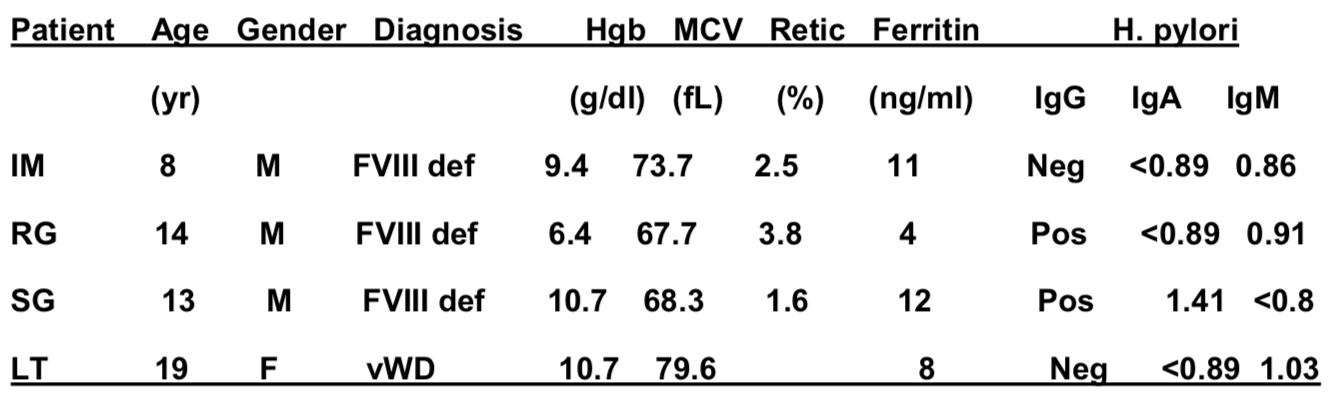
As of June 13, 2014, 25 HTCs were enrolling patients and 865 patients were enrolled. Of those patients, 168 opted for clinical genotyping only and 697 also gave informed consent to have data and samples entered into the research repository. Mutation analysis has been completed in 707 patients, including 354, 151 and 202 with severe, moderate and mild haemophilia respectively. By comparison to available hemophilia A and B databases, 61 novel mutations have been identified in 68 patients. Lessons learned from the initial stages of the program’s rollout helped us to improve our approach to recruitment and education about the importance of genotyping and research in hemophilia.
Conclusions:
My Life, Our Future is a novel partnership to address unmet needs for hemophilia genotyping services and research. Expanding participation in the program will increase clinical genotyping for patients with hemophilia, increase knowledge of FVIII and FIX mutations present in the U.S. hemophilia population, and provide a robust research repository for future scientific discovery.