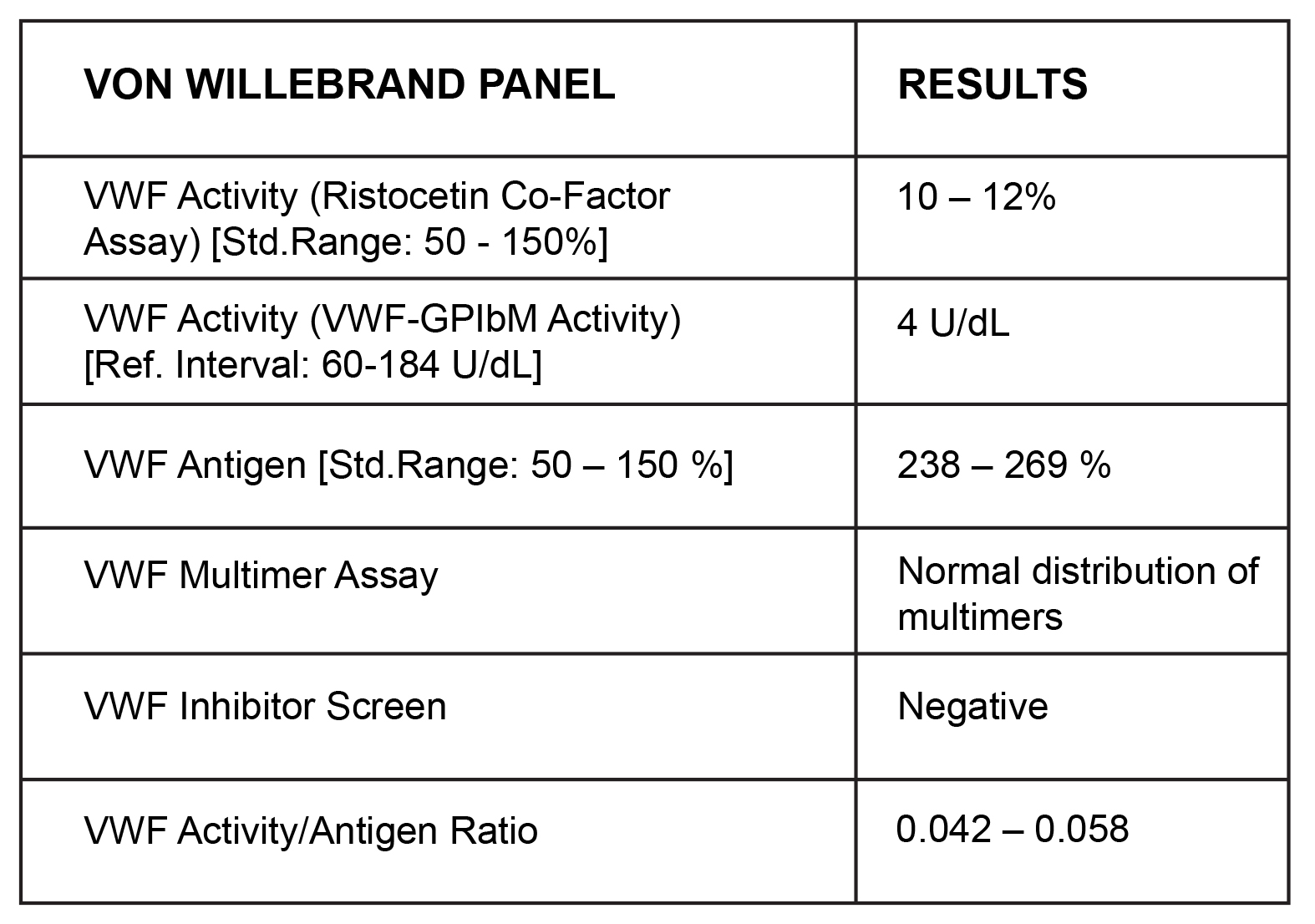
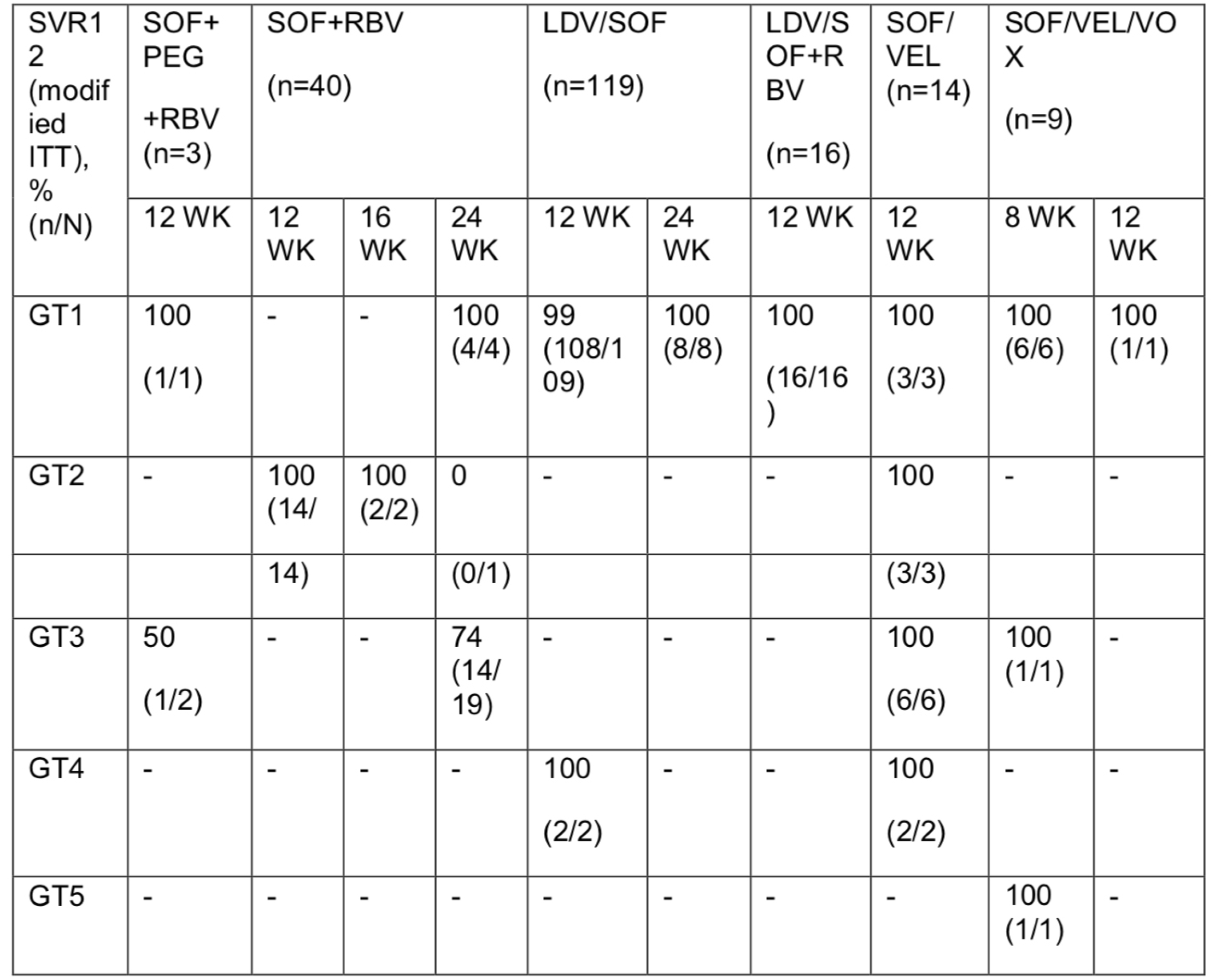
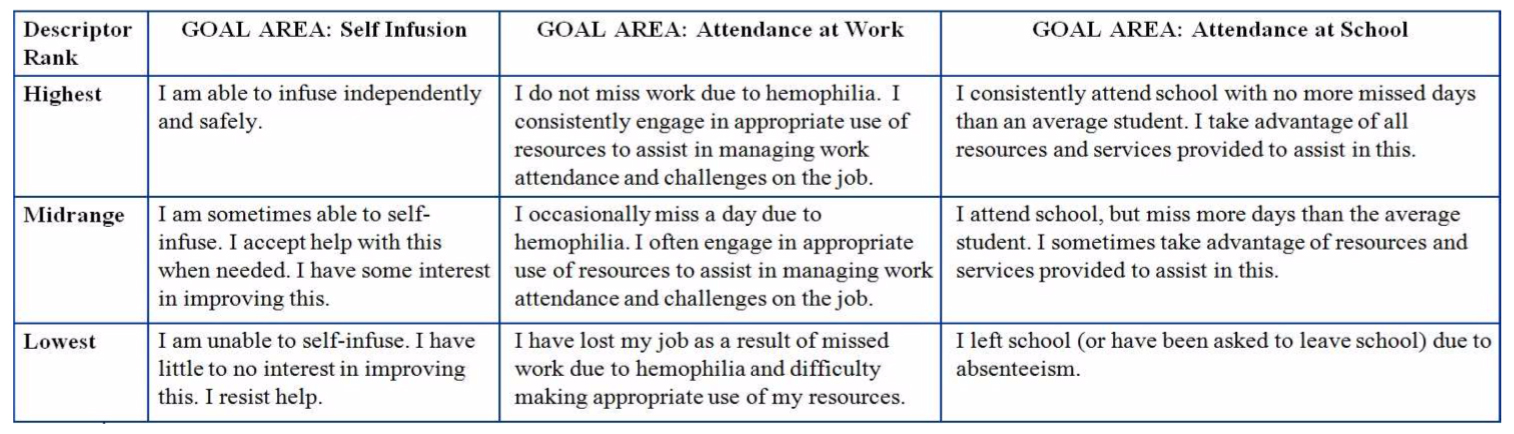
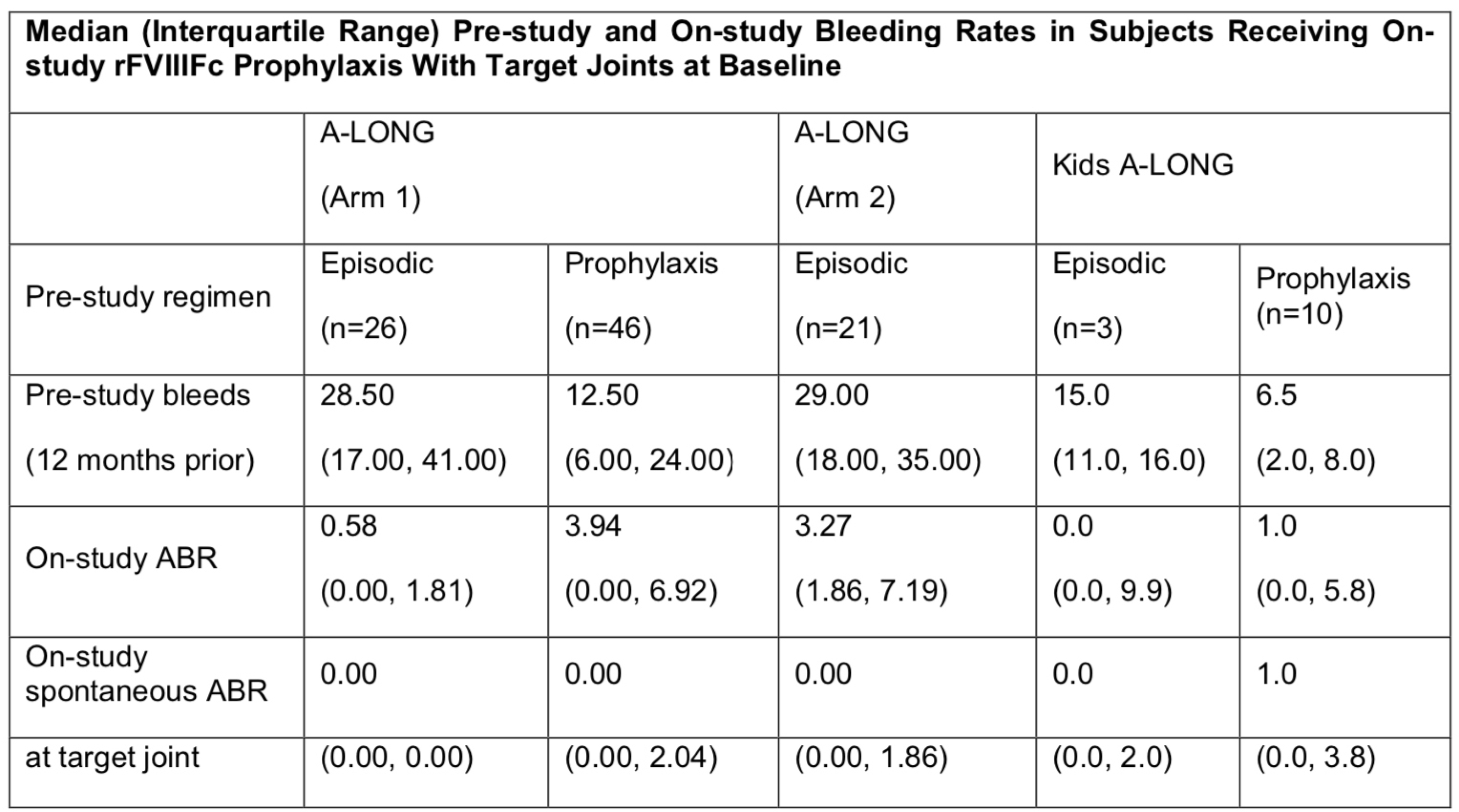
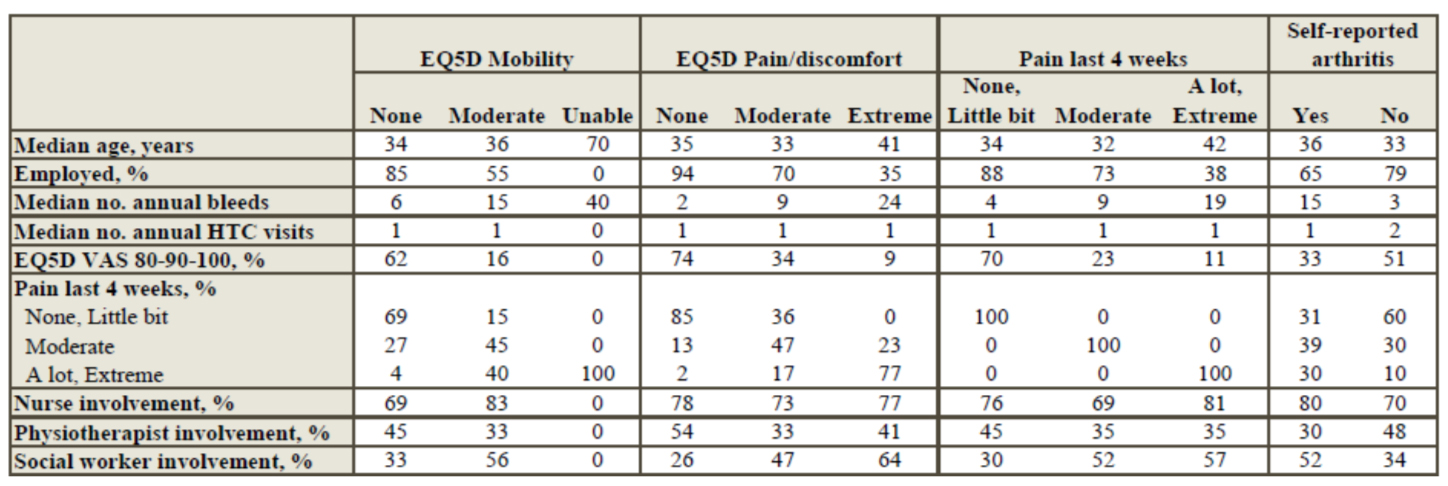
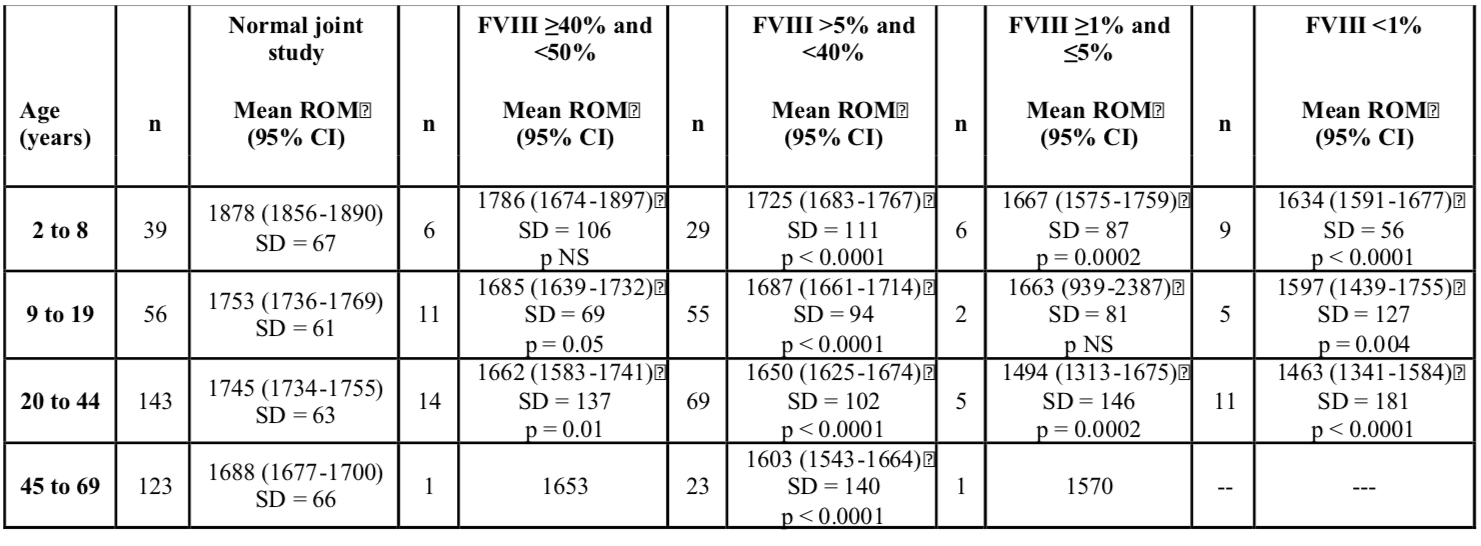
Introduction and Objectives:
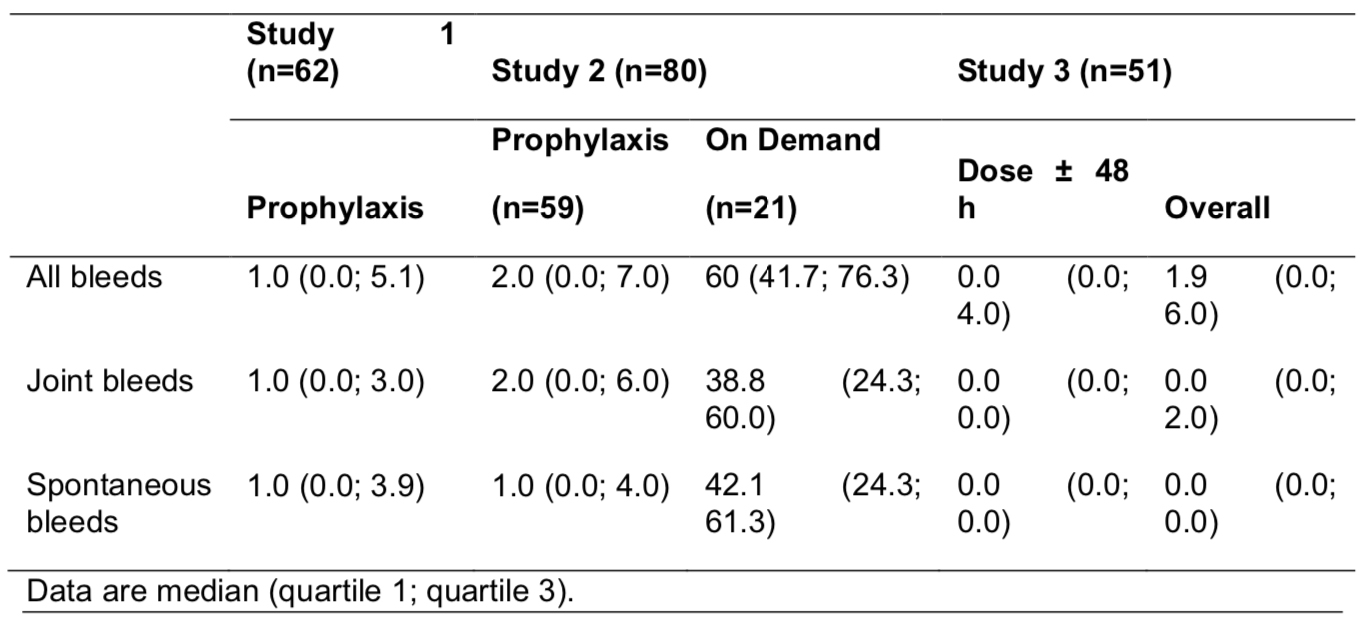
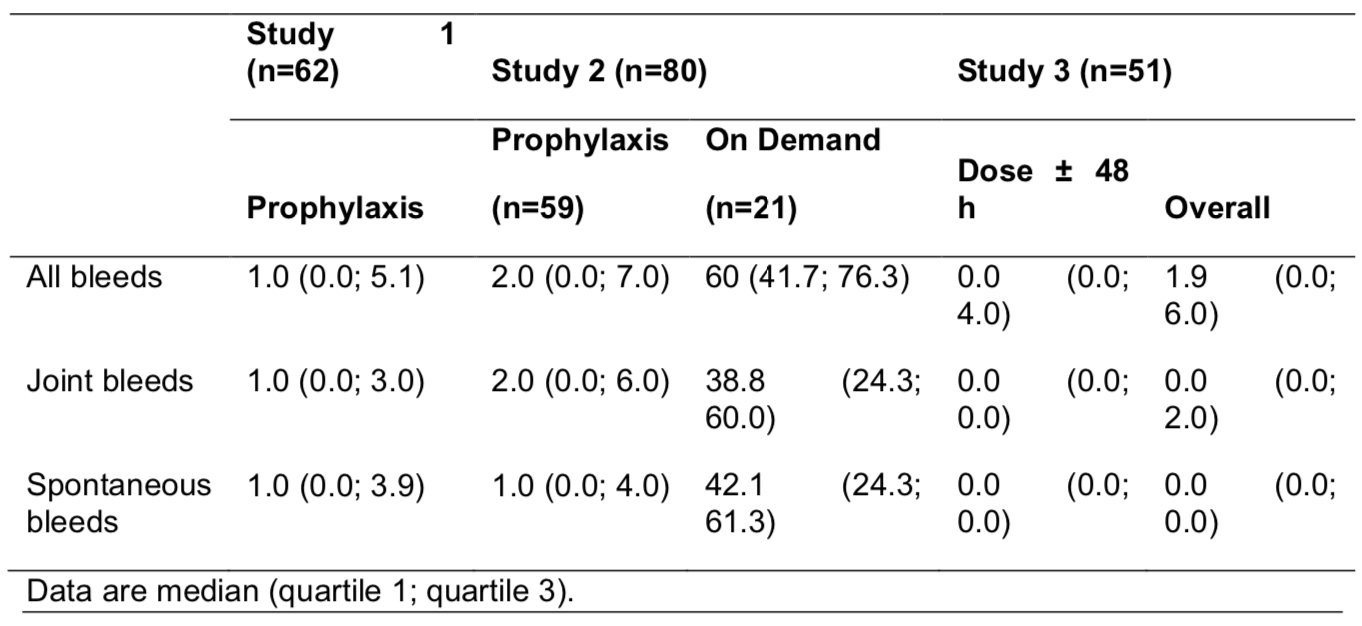
In the phase 3 A-LONG and Kids A-LONG studies, subjects with severe hemophilia A receiving rFVIIIFc prophylaxis 1-2 times/week had low annualized bleeding rates (ABRs), with comparable pre-study and on-study weekly factor consumption for subjects previously on FVIII prophylaxis. This report evaluated the effect of rFVIIIFc on subjects’ physical activity across a variety of age groups using a subject-reported assessment.
Methods:
Subjects eligible for A-LONG (≥12 y) and Kids A-LONG (<12 y) were previously treated males with severe hemophilia A (<1 IU/dL endogenous FVIII activity). Subjects in A- LONG were enrolled into 1 of 3 arms: Arm 1, individualized prophylaxis; Arm 2, weekly prophylaxis; or Arm 3, episodic treatment. All subjects in Kids A-LONG received rFVIIIFc prophylaxis. There were no restrictions regarding physical activity. Physical activity assessments were conducted at Weeks 7, 14, 28, 38, 52, and end of study (A-LONG) and Weeks 2, 7, 12, 17, 22, 26, and end of study (Kids A-LONG). At each visit after their first rFVIIIFc dose, subjects were asked to report any changes in their activity levels relative to their prior study visit as: more (or more intensive), fewer (or less intensive), or about the same amount of physical activities. To summarize each subject’s change in physical activity during the study compared to baseline, subjects’ reports were classified into four groups: less, the same, more, or undetermined.
Results:
A total of 165 and 71 subjects enrolled in A-LONG and Kids A-LONG, respectively. Overall, the majority of subjects in A-LONG reported more or the same amount of physical activity, and few subjects reported less physical activity during the study (less, the same, more, undetermined in Arm 1 [n=117], 8%, 36%, 51%, 5%; Arm 2 [n=23], 9%, 48%, 39%, 4%; Arm 3 [n=23], 9%, 52%, 26%, 13%, respectively). Results were generally similar for subjects in Kids A-LONG (for subjects aged <6 y [n=35], 3%, 26%, 66%, 6%; for subjects aged 6 to <12 y [n=34], 9%, 26%, 56%, 9%).
Conclusions:
The majority of subjects in A-LONG and Kids A-LONG reported similar or increased physical activity levels during the studies, while maintaining low ABRs. These self- reported data suggest that subjects across a variety of age groups with severe hemophilia A who are transitioning to rFVIIIFc may maintain or increase physical activity levels, while reducing infusion frequency and maintaining similar weekly factor consumption, without compromising efficacy.